


如题,最近要进行一些计算量很大的生信分析,登陆节点我怕影响其他人使用,想用作业系统提交到计算节点上,但是学校的使用手册并不是很清楚,想请教一些问题
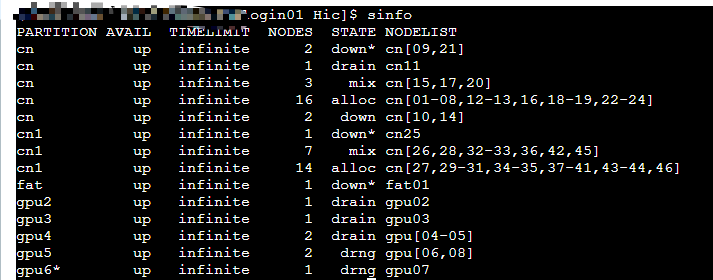 这些节点有哪些可用?根据手册似乎都被占用了或是不可用?
这些节点有哪些可用?根据手册似乎都被占用了或是不可用?

我的计算任务提交到 cpu 节点就可以,计算任务示例如下
pigz -p 24 -d genome_ucsc_mm39.fa.gz
bowtie2-build -f genome_ucsc_mm39.fa genome_ucsc_mm39.fa.bowtie2_index --threads 24
ls *.sra | parallel fastq-dump --gzip --split-3 {} --outdir ./fastqgz
ls ./fastqgz/*.fastq.gz | xargs fastqc -t 2 -O ./fastqc_reports
multiqc ./fastqc_report # 合并质量检测报告
############# Trim_galore filtering sequence #############
cat ./SRR_Acc_List.txt | while read id; do
nohup trim_galore \
-q 20 \
--length 36 \
--max_n 3 \
--stringency 3 \
--fastqc \
--paired \
-o ./clean/ \
./fastqgz/${id}_1.fastq.gz ./fastqgz/${id}_2.fastq.gz &
done
wait # 等待 trim_galore 完成
genome='../oryza_sativa.fa' # 基因组文件路径(需提前下载解压)
species='oryza_sativa' # 物种名称(作为索引名称)
cd ./genome_index
cp ${genome} ./
hisat2-build -p 4 ${species}.fa ${species}
cd ..
cat ./SRR_Acc_List.txt | while read id;
do
hisat2 \
-x ./genome_index/${species} \
-p 5 \
-1 ./clean/${id}_1.fastq.gz \
-2 ./clean/${id}_2.fastq.gz \
-S ./compared/${id}.sam
done
学校的超算使用手册在此处
应该如何合理地提交计算任务?
7 条回复 • 2024-04-04 21:55:14 +08:00
 |
1
ZedRover 28 天前
你直接提交任务到你想用的节点上,会排队的,排到了才会运行
|
 |
2
catscan 28 天前
时间长的写出脚本,用 sbatch 提交,手册里不是写清楚了吗
|
|
3
catscan 28 天前
把需要几个分区,几个核心数量设置好就可以了,分区是自动分配的,当然也可以指定分区使用,不过没必要
|
 |
4
tap91624 28 天前
看起来是 slurm 那一套其实大差不差
|
 |
5
TongDu 28 天前
下面是是一个例子,使用一个节点(-N 1 ),以及节点上的 24 个 CPU 核心数(-c 24 ),把下面的内容保存成一个单独文件,比如 submit.slurm 。然后在 shell 执行`sbatch submit.slurm`即可,提交后可以通过在 shell 输入`squeue`查看任务状态。如果想要了解更多,可以查看官方文档 https://slurm.schedmd.com/ 。也可以搜索 slurm 平台提交作业的教程,slurm 是一个开源的 HPC 平台,教程还是很多的。
``` #!/bin/bash #SBATCH -J myjob #SBATCH -o %j.job.out #SBATCH -e %j.job.error #SBATCH -N 1 #SBATCH -n 1 #SBATCH -c 24 #SBATCH -p cn pigz -p 24 -d genome_ucsc_mm39.fa.gz bowtie2-build -f genome_ucsc_mm39.fa genome_ucsc_mm39.fa.bowtie2_index --threads 24 ``` 另外我附上两个教程: https://docs.hpc.sjtu.edu.cn/job/slurm.html http://faculty.bicmr.pku.edu.cn/~wenzw/pages/slurm.html |

 |
7
abbottcn 25 天前
用户无需指定节点;
用户仅需申明,我需要几个处理器核心,是否限定在一个计算节点,每一个处理器核心配置多少内存。 这里任何一项,超出实际物理额度或者超过用户所在群组的资源限定上限,计算都是无法运行的。 用户如手动指定节点名称,计算可能无法运行或者超长时间等待。 一般的超算,默认限定计算任务在一个主板上进行;当然超算的硬件是支持多机器多核心并行的,不过也需要查验确认自己的应用,在多机器/跨节点运行的时候,所用的 MPI 是否支持 IB 网络。否则,由于超算上并发 I/O 频繁,可能导致 I/O 性能堪忧,进而导致您的计算运行效率低下。 |